

Brief overview of posterior reversible encephalopathy syndrome (PRES) and how it may present in the Emergency Department. This blog has been written and peer-reviewed by emergency physicians.
Posts tagged #critical care
Toxic Alcohols

Written by: Rafael Lima, MD (NUEM ‘23) Edited by: Laurie Aluce, MD (NUEM ‘21)
Expert Commentary by: Zachary Schmitz (NUEM ‘21)
Methanol Toxicity
Methanol itself is not toxic to the body. Methanol’s metabolite, formic acid, causes toxicity at serum levels greater than 20mg/dl [1].

Clinical Findings of Methanol Poisoning
CNS sedation
Seizures
Rapid, Deep Breathing
Hypotension
Ocular findings:
Blindness
Afferent pupillary defect
Optic disk hyperemia
Mydriasis
Ethylene Glycol Toxicity
Similarly, the toxic metabolites of ethylene glycol cause end-organ damage at levels greater than 20mg/dl. The most notable toxic metabolites are glycolic acid and oxalic acid.” [1] .

Clinical Findings of Ethylene Glycol Poisoning
CNS sedation
Seizures
Cranial nerve palsies
Rapid, deep breathing
Hypotension
Hypocalcemia (can result in tetany)
Renal findings:
Oliguria
Acute renal failure
Flank pain
Hematuria
Oxalate crystals in the urine under fluorescence
Isopropyl Alcohol Toxicity
Found in hand sanitizers and disinfectants, isopropyl alcohol is a less common source of alcohol poisoning. The parent molecule does exhibit toxic effects here, unlike methanol and ethylene glycol. If untreated, the lethal dose is between 4-8 g/kg [2].
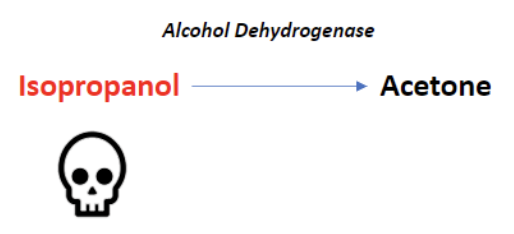
Alcohol dehydrogenase metabolizes isopropyl alcohol into acetone. Because acetone is a ketone, and ketones are not oxidized into carboxylic acids, isopropyl alcohol poisoning does not result in anion gap metabolic acidosis.
Clinical Findings of Isopropyl Alcohol Poisoning
CNS sedation
Disconjugate gaze
Fruity breath odor
Hypotension
Hematemesis
Pulmonary edema
Plasma Osmolal Gap
One of the most reliable laboratory markers of toxic alcohol poisoning is a large osmolal gap. The osmolal gap is defined as the difference between the measured serum osmolality and the calculated, or expected, plasma osmalality:
OSMOLAR GAP = Measured plasma osmolality – calculated/expected plasma osmolality
The common equation for calculating the expected plasma osmolality is listed below [3]. Of note, there are other formulas with slight variations. Using an online calculator can be helpful.
Expected Serum Osmolality=2[Na]+BUN/2.8+Glucose/18
A gap < 10 is considered normal. Any elevation above 10 should raise the clinician’s suspicion of toxic alcohol ingestion.
Note: this tool is not helpful in late presentations as the metabolized forms of the different alcohols do not contribute to the osmolal gap. The calculated gap will be falsely low in late-stage poisoning.
Treatment of Toxic Alcohol Ingestions
Consult your medical toxicologist or poison control center if toxic alcohol ingestion is suspected.
The national poison control center hotline telephone number is 1(800)-222-1222.
Fomepizole
Fomepizole should be used only for methanol and ethylene glycol ingestions. It is not indicated for isopropyl alcohol intoxications [4]. It is an inhibitor of alcohol dehydrogenase (ADH). Evidence shows that it is a superior antidote to ethanol [5].
Loading dose 15 mg/kg IV
Then 10 mg/kg every 12 hours
Continue until blood pH is normal and serum alcohol concentration is less than 20 mg/dL in the presence of retinal or renal injury.
Ethanol
Ethanol works as a competitive inhibitor of ADH, having a higher affinity for the enzyme compared to the other alcohols. Ethanol was used historically before the effects of fomepizole were studied. Fomepizole is now the preferred treatment because the administration of ethanol is more difficult, ethanol causes sedation, and titration of the therapy is challenging in co-ingestions [6]. If ethanol must be used, the preferred route is IV and the studied therapeutic target level is 100 mg/dL [7].
Supplemental Therapy
Methanol poisoning patients should also receive folic acid (50mg IV every 6 hours) [7].
Ethylene glycol poisoning patients should also receive thiamine (100mg IV) and pyridoxine (50mg IV) [8].
Hemodialysis
Consult your nephrologist early if you are considering hemodialysis. Renal replacement therapy should be considered in the following situations [9]:
Anion gap metabolic acidosis with known toxic alcohol ingestion
End-organ damage
Renal failure
Vision changes
Unexplained anion gap metabolic acidosis with elevated osmolal gap in suspected toxic alcohol ingestion
References
1. Liesivuori, J. and H. Savolainen, Methanol and formic acid toxicity: biochemical mechanisms. Pharmacol Toxicol, 1991. 69(3): p. 157-63.
2. Slaughter, R.J., et al., Isopropanol poisoning. Clin Toxicol (Phila), 2014. 52(5): p. 470-8.
3. Bhagat, C.I., et al., Calculated vs measured plasma osmolalities revisited. Clin Chem, 1984. 30(10): p. 1703-5.
4. Su, M., R.S. Hoffman, and L.S. Nelson, Error in an emergency medicine textbook: isopropyl alcohol toxicity. Acad Emerg Med, 2002. 9(2): p. 175.
5. McMartin, K., D. Jacobsen, and K.E. Hovda, Antidotes for poisoning by alcohols that form toxic metabolites. Br J Clin Pharmacol, 2016. 81(3): p. 505-15.
6. Zakharov, S., et al., Fomepizole versus ethanol in the treatment of acute methanol poisoning: Comparison of clinical effectiveness in a mass poisoning outbreak. Clin Toxicol (Phila), 2015. 53(8): p. 797-806.
7. Barceloux, D.G., et al., American Academy of Clinical Toxicology practice guidelines on the treatment of methanol poisoning. J Toxicol Clin Toxicol, 2002. 40(4): p. 415-46.
8. Ghosh, A. and R. Boyd, Leucovorin (calcium folinate) in "antifreeze" poisoning. Emerg Med J, 2003. 20(5): p. 466.
9. Moreau, C.L., et al., Glycolate kinetics and hemodialysis clearance in ethylene glycol poisoning. META Study Group. J Toxicol Clin Toxicol, 1998. 36(7): p. 659-66.
Expert Commentary
Thank you for this great review of a difficult subject! The combination of a lack of quick, confirmatory testing with delayed onset of symptoms makes toxic alcohol poisoning an incredibly difficult diagnosis to make. Additionally, even small ingestion can lead to major complications. For example, if a typical four-year-old (19kg) child drank windshield washer fluid that contained 50% methanol (a fairly standard formulation), it would take only 5.7 mL to potentially produce a methanol serum concentration of 25 mg/dL. Given the average 4-year-old’s mouthful is 8.9 mL, you can run into trouble quickly.[1]
We frequently see misuse or misunderstanding of osmol and anion gaps in diagnosing toxic alcohol ingestion when history is unclear. First, although a normal osmol gap is generally less than 10, baseline osmol gaps range from -10 to +14.[2] Therefore, a gap of 16 may represent a true gap of +2 in one person and +26 in another. Second, ethanol must be included in the osmol gap equation. An ethanol concentration of 200 mg/dL would increase your osmol gap by 43.5. Third, given metabolism over time, all values included in an anion gap calculation need to be drawn off of the same blood sample.
These considerations make finding the diagnosis even more complicated, but there are a few things that can help you out. First, an osmol gap > 50 is highly concerning for toxic alcohol. Second, an ethanol concentration > 100 mg/dL is sufficient to block ADH, meaning that few toxic metabolites from methanol or ethylene glycol could be made.[3] This means that an anion gap present with an ethanol > 100 mg/dL is not from toxic alcohol (unless the patient drank the ethanol after the toxic alcohol, which is very rare). Third, sequential values over time can be helpful. Metabolism of toxic alcohols should lead to a decreased osmol gap and increased anion gap over time. Proper use of the osmol and anion gap can help identify patients at high risk for morbidity and mortality while decreasing unnecessary administration of fomepizole, which typically costs thousands of dollars.
References
Ratnapalan S, Potylitsina Y, Tan LH, Roifman M, Koren G. Measuring a toddler's mouthful: toxicologic considerations. Journal of Pediatrics. 2003 Jun;142(6):729-30. doi: 10.1067/mpd.2003.216
Hoffman RS, Smilkstein MJ, Howland MA, Goldfrank LR. Osmol gaps revisited: normal values and limitations. J Toxicol Clin Toxicol. 1993;31(1):81-93. doi: 10.3109/15563659309000375.
Jacobsen D, McMartin KE. Methanol and ethylene glycol poisonings: mechanism of toxicity, clinical course, diagnosis and treatment. Med Toxicol. 1986;1:309-334.

Zachary Schmitz, MD
Toxicology Fellow
Ronald O. Perelman Department of Emergency Medicine
NYU Langone Health
How To Cite This Post:
[Peer-Reviewed, Web Publication] Lima, R. Aluce, L. (2022, Jan 24). Toxic Alcohols. [NUEM Blog. Expert Commentary by Schmitz, Z]. Retrieved from http://www.nuemblog.com/blog/toxic-alcohols
Other Posts You May Enjoy








Crashing Patient on a Ventilator
Written by: Patrick King, MD (NUEM ‘23) Edited by: Adesuwa Akehtuamhen, MD (NUEM ‘21)
Expert Commentary by: Matt McCauley, MD (NUEM ‘21)

Expert Commentary
Thank you for this succinct summary of an incredibly important topic. We as emergency physicians spend a lot of time thinking about peri-intubation physiology but the challenges do not end once the plastic is through the cords. The frequency with which our ventilated patients stay with us in the ED has been increasing for years and will likely continue to do so1. This means that managing both acute decompensation and refractory hypoxemia needs to be in our wheelhouse.
The crashing patient on the ventilator can be truly frightening and your post effectively outlines a classic cognitive forcing strategy for managing these emergencies. A truism in resuscitation is to always rule out the easily correctable causes immediately. In this case, it means removing the complexity of the ventilator and making things as idiot-proof as possible. Once you’ve ruled out the life threats like pneumothorax, tube displacement, and vent malfunction, you can try to bring their sats up by bagging. Just make sure that you have an appropriately adjusted PEEP valve attached to your BVM for your ARDS patients; the patient who was just requiring a PEEP of 15 isn’t going to improve with you bagging away with a PEEP of 5.
Once you’ve gotten the sats up and the patient back on the vent, your ventilator display can provide you with further data as to why your patient decompensated. Does the flow waveform fail to reach zero suggesting breath stacking and a need for a prolonged expiratory time? Is the measured respiratory rate much higher than your set rate with multiple breaths in a row indicating double-triggering? The measured tidal volume might fall short of your set tidal volume. This points towards a circuit leak, cuff leak, or broncho-pleural fistula. Maybe you’re seeing the pressure wave dip below zero mid-inspiration and the patient is telling you that they are in need of faster flow, a bigger breath, or deeper sedation. In these situations, your respiratory therapist is going to be your best friend in managing this patient-ventilator interactions2.
As your post alludes to, sometimes patients remain hypoxemic despite our usual efforts and refractory hypoxemia can be an intimidating beast when you’ve got a busy ED burning down around you. If your cursory efforts to maintain vent synchrony by playing with the ventilator dials have failed, there’s no shame in deepening sedation which will work to decrease oxygen consumption and prevent derecruitment. Once sedated, work with your RT to find appropriate PEEP and tidal volumes to meet your goals.
Most patients can be managed with usual lung-protective ventilation but some patients will require more support and you’ve correctly identified several salvage therapies. My general approach is to pursue prone positioning in any patient with a P:F ratio approaching 150 despite optimal vent settings as it has the only strong mortality benefit of the therapies outlined above. Proning in the ED is resource intensive and is probably better pursued as a department-wide protocol rather than you and your charge nurse trying to figure it out in the middle of the night3.
As you’ve pointed out, the neuromuscular blockade has more limited evidence and is not required for prone ventilation. Upstairs, we accomplish this with continuous infusions but in the ED you may be more comfortable using intermittent boluses of intubation dose rocuronium. Just make sure your patient is unarousable. I reach for this if I’m unable to achieve ventilator synchrony with sedation alone as it allows for very low tidal volumes and inverse ratio ventilation. I see inhaled pulmonary vasodilators in a similar light: there’s no data on patient-oriented outcomes but they can make your numbers look prettier while you wait for more definitive interventions such as transfer.
This finally brings me to VV ECMO for refractory hypoxemia. It’s worth considering that while there is some evidence for a mortality benefit for ECMO in ARDS, the evidence base is mixed. The CESAR trial did show a mortality benefit in patients transferred to an ECMO center but only 76% of patients actually received ECMO upon transfer4. The larger and more recent EOLIA trial failed to demonstrate this improvement in mortality5. The conclusion I take from this is that treatment at a high volume center matters and that a boarding patient with refractory hypoxemia warrants an early consideration for transfer to a tertiary center if high-quality ARDS care can’t be accomplished upstairs at your shop.
References
Mohr NM, Wessman BT, Bassin B, et al. Boarding of Critically Ill Patients in the Emergency Department. Crit Care Med. 2020;48(8):1180-1187. doi:10.1097/CCM.0000000000004385
Sottile PD, Albers D, Smith BJ, Moss MM. Ventilator dyssynchrony – Detection, pathophysiology, and clinical relevance: A Narrative review. Ann Thorac Med. 2020;15(4):190. doi:10.4103/atm.ATM_63_20
McGurk K, Riveros T, Johnson N, Dyer S. A primer on proning in the emergency department. J Am Coll Emerg Physicians Open. 2020;1(6):1703-1708. doi:10.1002/emp2.12175
Peek GJ, Mugford M, Tiruvoipati R, et al. Efficacy and economic assessment of conventional ventilatory support versus extracorporeal membrane oxygenation for severe adult respiratory failure (CESAR): a multicentre randomised controlled trial. Lancet Lond Engl. 2009;374(9698):1351-1363. doi:10.1016/S0140-6736(09)61069-2
Combes A, Hajage D, Capellier G, et al. Extracorporeal Membrane Oxygenation for Severe Acute Respiratory Distress Syndrome. N Engl J Med. Published online May 23, 2018. doi:10.1056/NEJMoa1800385

Matt McCauley, MD
How To Cite This Post:
[Peer-Reviewed, Web Publication] King, P. Akehtuamhen, A. (2022, Feb 28). Crashing Ventilator Patient. [NUEM Blog. Expert Commentary by McCauley, M]. Retrieved from http://www.nuemblog.com/blog/crashing -vent-patient.
Other Posts You May Enjoy




Vasopressor Nonresponse

Written by: Elizabeth Stulpin, MD (NUEM ‘23) Edited by: Aaron Wibberly, MD (NUEM ‘22)
Expert Commentary by: Joshua Zimmerman, MD (NUEM ‘17)
Non-Response to Vasopressors
Shock is defined as a state of cellular and tissue hypoxia resulting in end organ dysfunction. This state may arise due to impaired oxygen delivery to tissues, impaired oxygen utilization by the tissues themselves, increased oxygen consumption, or a combination of these mechanisms. Due to its extremely high morbidity and mortality as well as high healthcare costs, the prompt recognition, diagnosis and resuscitation of shock is key. And for most forms, EM physicians are not typically shocked by shock. They have a toolbox of strategies, mainly fluids and vasopressors, to stabilize these critically ill patients.
However, what happens when the trusted treatment paradigm fails? There is a subset of patients who, despite aggressive conventional resuscitation, have an inadequate hemodynamic response and develop refractory shock. This is seen in approximately 7 percent of patients, with short-term mortality ranging from 50 to 80 percent. Due to this significantly lower incidence and increased mortality, alternate causes for refractory shock must be considered when vasopressors do not have the desired effect.
Acidosis
Acidosis in shock states can present from multiple different sources, including sepsis, hypoxemia, ingestions, hyperlactatemia from hypoperfusion, amongst others. With increasing acidosis, calcium influx is reduced, contractility is inhibited and the binding affinity of pressors is reduced, all of which lead to excess vasodilation and refractory hypotension. While bicarbonate is sometimes given in an effort to increase cellular pH, it is controversial for any pH >7.0. At those levels, bicarbonate administration has not been shown to improve cardiac output, MAP or pressor response. While a bicarbonate drip and hyperventilation can temporize an acidosis, emergent HD or CRRT is a definitive treatment if the cause cannot be quickly reversed.
Adrenal Insufficiency
Cortisol has a myriad of functions in the body, not limited to its synergistic effects with catecholamines to help cause vasoconstriction. Thus, when the adrenal glands are chronically suppressed and then experience an acute stressor, hypotension can ensue. The most common cause of chronic suppression is long-term steroid use, and a stressor can include surgery, infection, hypovolemia, pregnancy, medications, or reduced steroid use. Clues that suggest adrenal insufficiency include nausea/vomiting, cutaneous hyperpigmentation, and multiple electrolyte abnormalities (hyponatremia, hyperkalemia, hypoglycemia). Previously healthy individuals, in the setting of critical illness, can also infrequently decompensate into a state of relative adrenal insufficiency. To reverse these effects as well as refractory hypotension, hydrocortisone is the preferred agent due to both its glucocorticoid and mineralocorticoid properties. A loading dose of 100mg IV should be given, followed by 50mg every 6 hours thereafter.
Alternate Shock
Not all shock is declared equal. For example, a patient in cardiogenic shock will likely worsen with the administration of fluids and the wrong vasopressors. Similarly, obstructive shock as seen in massive PEs, tension pneumothoraces or cardiac tamponade will not improve without addressing the cause.
Anaphylaxis
Anaphylaxis may present as hypotension alone. Thus, it may easily be confused with a different form of shock and treated with vasopressors such as norepinephrine and vasopressin, which are not first line for anaphylaxis. Along with using epinephrine as the pressor of choice and other conventional therapies for anaphylaxis, there are alternate medications available for persistent refractory hypotension. One of these is methylene blue. While typically reserved for treatment of methemoglobinemia, the cellular mechanism of methylene blue can decrease vasodilation. Data suggest that this effect can be seen with a one-time dose of 1-2mg/kg.
Hemorrhage
Often a common cause of refractory shock in the post op setting, bleeding can be obscure in its early stages before a hemoglobin drop is appreciated or before the patient develops abdominal distension and flank dullness (retroperitoneal bleed). If concerned, empiric uncrossed unmatched blood can be transfused.
Hypocalcemia
Calcium homeostasis is necessary for the proper maintenance of myocardial contractility and vascular tone. Hypocalcemia can be hinted at through history or by hints such as a prolonged QTc on an ECG. Those at higher risk of hypocalcemia (vitamin D deficiency, ESRD, hyperparathyroidism, burns, multiple blood transfusions, etc.) may have greater severity of shock with increased mortality. In repleting calcium, co-administration of phosphorous and magnesium may allow for reversal of the patient’s shock state.
Hypothyroidism
Decompensated hypothyroidism can have profound effects, including bradycardia, impaired myocardial contractility, and decreased peripheral vasoconstriction. However, even subclinical hypothyroidism with an elevated TSH but normal T4 increases risk of poor outcomes due to effects on cardiac function. While the diagnosis of hypothyroidism can be delayed by lab results, clues to the diagnosis include thyroidectomy scar, non-pitting edema of the extremities, macroglossia, altered mental status, hypoglycemia and hypothermia. Initial hypotension may not respond to vasopressors, but shock should improve once thyroid hormone is given. Stress dose steroids (hydrocortisone 100mg IV) should also be given due to the association between adrenal insufficiency and hypothyroidism, and giving thyroid hormone without steroids can precipitate adrenal crisis.
Ingestions
When in doubt, look at the medication list! Both beta blocker and calcium channel blocker toxicity can cause profound myocardial depression, bradycardia and hypotension due to their inhibition of calcium signaling. Refractory hypotension can be overcome with the use of direct cardiac pacing, calcium, glucagon, or high dose insulin. And if all else fails, ECMO can overcome medication toxicity until it can be fully metabolized or cleared.
When conventional resuscitation for shock with fluids, vasopressors and/or inotropes fails, it is time for a cognitive pause. By running through this list of alternate causes of refractory shock, other methods of resuscitation can be added to improve patient outcomes and stabilize the patient.
Sources:
1) Amrein K, Martucci G, and Hahner S. Understanding adrenal crisis. Intensive Care Medicine. 2018; 44(5): 652-655.
2) Boyd J, Walley K. Is there a role for sodium bicarbonate in treating lactic acidosis from shock? Curr Opin Crit Care. 2008;14:379-83.
3) Farkas J. Decompensated Hypothyroidism. The Internet Book of Critical Care. 2016. Accessed https://emcrit.org/ibcc/myxedema/
4) Ho H, Chapital A, and Yu M. Hypothyroidism and Adrenal Insufficiency in Sepsis and Hemorrhagic Shock. Arch Surgery. 2004; 139(11):1199-1203.
5) Kerns W. Management of B-Adrenergic Blocker and Calcium Channel Antagonist Toxicity. Emergency Medicine Clinics of North America. 2007; 25: 309-331.
6) Levy B, Collin S, Sennoun N, et. al. Vascular hyporesponsiveness to vasopressors in septic shock: from bench to bedside. Intensive Care Medicine. 2010; 36: 2019-2029.
7) Manji F, Wierstra B, and Posadas J. Severe Undifferentiated Vasoplegic Shock Refractory to Vasoactive Agents Treated with Methylene Blue. Case Reports in Critical Care. 2017.
8) Minisola, S et al. Serum Calcium Values and Refractory Vasodilatory Shock. Chest. 2019; 155(1): 242.
9) Nandhabalan P, Ioannou N, Meadows C and Wyncoll D. Refractory septic shock: our pragmatic approach. Critical Care. 2018; 22(1):215.
10) Smith L, and Branson B. Refractory Hypotension – Diagnosis and Management in Surgical Patients. California Medicine. 1961; 95(3): 150-155
11) Velissaris D, Karamouzos V, Ktemopoulos N, Pierrakos C, and Karanikolas M. The Use of Sodium Bicarbonate in the Treatment of Acidosis in Sepsis: A Literature Update on a Long Term Debate. Critical Care Research and Practice. 2015.
12) Wang H, Jones A, and Donnelly J. Revised National Estimates of Emergency Department Visits for Sepsis in the United States. Critical Care Medicine. 2017; 45(9): 1443-1449.
Expert Commentary
Dr. Stulpin's review of a very critical topic is well articulated and concise. I would like to particularly emphasize her final points before delving into the details. As she pointed out, patients failing to respond to typical resuscitative efforts represent quite a quagmire to the ED physician. It is easy to arrive at premature closure and presume all shock to be sepsis simply needing more fluids or vasopressors. However, there is significant risk in this practice, and I would thus advocate very strongly for a “pause” and thorough reassessment of the patient’s presentation and condition. Much akin to a pre-procedural time out, this should be deliberate and uninterrupted to prevent diagnostic momentum from building and arriving at a premature closure on the etiology of the patient’s condition.
This review covers much of the differential and pathophysiology that is germane to a discussion of refractory shock. Rather than review this in detail, I would like to discuss a practical approach to application of this knowledge at the bedside. So, as we approach the patient who is refractory to standard resuscitation, one’s first task is to confirm the patient is receiving the desire therapy and that this is truly shock refractory to intervention. Check all access points for infiltration –the single most reversible cause for refractory hypotension is that the patient is not receiving fluids/vasopressors, etc. due to inadequate access. Check dosing and confirm with nursing that infusions have been running appropriately. If access is the issue, I would advocate placing a more reliable form such as central access or, in the very unstable, IO access early in the reassessment. While I am an advocate for US guided access in stable patients and feel that this is often a great tool, this is a scenario I would advocate against US guided peripheral access. This form of access is often time consuming, and more importantly, signs of infiltration are less evident in deeper IV sites which is a serious concern if you are running peripheral vasopressors.
Presuming your access is adequate, the next task is to reassess perfusion in its entirety. While not mandatory, this is a point where I would consider placing an arterial line for optimally reliable BP monitoring. One must perform a complete re-examination as well. If not already completed, a rectal exam for the occult GI bleeding. One should additionally particular attention on reassessment to skin and perfusion. Check capillary refill, skin temperature and coloration as a matter of habit for cases of refractory shock. Cool extremities are not typically associated with distributive shock and should make you consider shock states with high SVR such as cardiogenic or acute blood loss. In addition, a repeat skin exam may reveal new rashes – I look specifically for the presence of petechiae, purpura or urticaria. Urticaria should prompt consideration of anaphylaxis. If the patient has received any antibiotics yet this is an important consideration given antibiotics are one of the most common medication precipitants of anaphylaxis and this may be the source of their worsening shock state.
After completing your thorough exam, I strongly advocate for a sonographic assessment as well. This is where the RUSH exam, or at least a modified version, fits well into patient assessment and can offer a great deal of information. I pay particularly close attention to the cardiac and abdominal windows. This can aid in the rapid diagnosis of an obstructive shock state such as cardiac tamponade, or acute RV failure in the setting of massive PE. Free intra-abdominal fluid should be considered hemorrhage until proven otherwise in the shock patient. One reminder is that when assessing for occult bleeding, the FAST exam views are an excellent tool but not sufficiently sensitive for definitive rule out. In particular, the FAST exam lacks sensitivity for retroperitoneal bleeding and therefore if there remains high clinical concern, CT imaging should also be considered.
Once a thorough re-examination is completed, reconsider the patient’s medications as another source. Patients are on a myriad of antihypertensives and other agents that can lower blood pressure. Many of the pathways that metabolize or eliminate these drugs are compromised in a state of hypoperfusion and can lead to a synergistic worsening of hypotension. One example of this is the case of BRASH Syndrome. In addition to antihypertensives, a review of the patient’s medication list should include a particular search for steroids or other adrenal replacements such as fludrocortisone. An extremely important cause of refractory hypotension to consider is that of adrenal insufficiency. As Dr. Stulpin reviewed in her discussion above, this can come in both primary and secondary forms. The latter is far more common and induced by exogenous steroid use. A wise ICU attending once taught that no patient should die without stress dose steroids. While perhaps a bit morbid, the take home point here was that adrenal insufficiency can present in any patient, not just those with underlying disorders of glucocorticoid production. Pay particular attention to patients with autoimmune disorders or others on chronic steroid therapy as these are the populations that are particularly at risk. Patients presenting with unexplained hypoglycemia in the setting of sepsis or shock of any kind should also be strongly considered to receive stress dose steroids. Do not wait for a cortisol level to treat this condition. Consider, as well, checking TSH and free T3/T4 as patients with adrenal insufficiency can simultaneously harbor other endocrinopathies.
To summarize, refractory hypotension or failure to respond to traditional interventions is relatively uncommon but critical to identify in shock patients. Often these patients have a primary diagnosis of septic shock but can suffer from concurrent shock related to one or more of the differential considerations we have reviewed above. A thoughtful reassessment, both of the patient’s physical exam findings, US and other diagnostics, medication list and history of present illness will offer clues that may uncover an additional etiology critical to treat and to ensure the best outcome possible.

Joshua D. Zimmerman, MD
Health System Clinician
Feinberg School of Medicine
Northwestern Medicine
How To Cite This Post:
[Peer-Reviewed, Web Publication] Stulpin, E. Wibberly, A. (2022, Jan 24). Vasopressor Nonresponse. [NUEM Blog. Expert Commentary by Zimmerman, J]. Retrieved from http://www.nuemblog.com/blog/vasopressor-nonresponse
Other Posts You May Enjoy






The BICAR-ICU Trial and Practical Use of Bicarb in Metabolic Acidosis

Written by: Philip Jackson, MD (NUEM ‘20) Edited by: Katie Colton, MD (NUEM ‘19) Expert Commentary by: Benjamin Singer, MD
Introduction
Until recently, there has been a paucity of high-quality data to inform the use of intravenous sodium bicarbonate in severe metabolic acidosis. This has resulted in a lack of universal practice guidelines to inform clinicians in emergency medicine and other specialties when caring for some of their sickest patients.
Historically there have been two camps of thought when approaching the use of sodium bicarbonate in the sick, acidotic patient. Severe acidemia results in protein dysfunction, potentially leading to arrhythmia, cardiovascular collapse, multi-organ failure and eventual death. [1-6] Thus, correcting acidemia with alkalotic bicarbonate solutions could prevent these compounding complications. However, growing evidence suggests that the deleterious effects associated with profound acidemia may be more strongly associated with the underlying physiological insult than the acidosis itself. Therefore, treating the acidemia without addressing the underlying pathology may expose the patient to side effects including hypernatremia, hypocalcemia, and exacerbation of CNS cellular acidosis (due to increased levels of carbon dioxide) without resulting in a net benefit. [1,2]
The BICAR-ICU Trial
A recent randomized, prospective, multi-center trial by Jaber et al. evaluated the effects of bicarbonate administration to ICU patients with metabolic acidemia. The study randomized 389 ICU patients with severe metabolic acidemia (pH ≤7⋅20, PaCO ≤45 mm Hg, and sodium bicarbonate concentration ≤20 mmol/L), a total Sequential Organ Failure Assessment score of 4 or more or an arterial lactate concentration of 2 mmol/L or more into a treatment group receiving 4.2% sodium bicarbonate (<1L per day) or a control group receiving an equivalent volume of standard crystalloid solution.
There was no significant difference between the two groups in the primary outcome, a composite of all-cause mortality at day 28 and the presence of organ failure at day 7. Interestingly, bicarbonate administration decreased the need for renal replacement therapy (RRT) and in a sub-group of patients with acute kidney injury bicarbonate infusion improved mortality and decreased vasopressor requirements.
It is important to note that the study excluded patients with significant urinary or digestive tract losses of bicarbonate (two important causes of non-anion gap metabolic acidosis) and patients that had already been treated with bicarbonate or RRT. Furthermore, a significant proportion of patients in the control group (24%) received bicarbonate at some point during the study and only 60% of the treatment group actually maintained the goal pH of >7.30. These factors skew the study towards a negative outcome, as they presumably blunt the effects of administration of bicarbonate. Thus, it is possible that more of a benefit may have been observed without these disruptions.
The study did not differentiate between etiologies of metabolic acidemia, though ketoacidosis was also excluded, and thus it is difficult to draw conclusions on the value of bicarbonate in various pathologic conditions. There was no specific protocol regarding timing of administration or concentration, making the study difficult to replicate in the emergency setting. Nevertheless, it was essentially the first large, multi-center RCT evaluating this topic and so some practical conclusions can be drawn; these should be interpreted in the context of each individual patient.
Practice recommendations in special situations
Anion Gap Metabolic Acidosis: The human body maintains an essentially neutral net electrical charge through the retention and excretion of ions (notably H+) and anions (notably Cl- and HCO3-). Addition or retention of other anions will increase the anion gap and cause a net negative charge, causing retention of H+ ions and leading to a metabolic acidosis. In general, administration of bicarbonate to this scenario will balance the pH but will not remove the additional anions that are the root cause of the pathologic acidosis and would presumably provide little benefit to patient outcomes.
Lactic Acidosis: Previous to the BICAR-ICU trial, most available data suggests no benefit of bicarbonate. Notably two small prospective physiological studies of 14 and 10 patients, respectively, demonstrated no hemodynamic response or difference in response to catecholamines. [7,8] Additional retrospective and observational studies did not result in clear conclusions. [9,10] The BICAR-ICU trial did not specifically evaluate this population and thus current guidelines recommend against bicarbonate administration unless pH falls below 7.15 or bicarbonate falls below 5 mEq/L (at which point small changes in bicarbonate concentration can lead to potentially fatal changes in pH). [11]
In general, DON’T GIVE IT
Diabetic and Alcoholic Ketoacidosis: A prospective RCT of 21 patients in severe DKA showed no benefit of bicarbonate therapy. [12] Limited data in pediatric DKA and adult AKA populations show similar findings. [13,14] There is evidence that bicarb administration is associated with worse outcome in pediatric patients. It is feasible to give bicarbonate to patients in extremis (pH<6.9) in the adult population to theoretically prevent cardiovascular collapse.
In general, DON’T GIVE IT except when pH<6.9 and with extreme caution in the pediatric patient
Toxic Ingestions (methanol, ethylene glycol, toluene, salicylates, etc.): In general, along with specific therapies, bicarbonate infusion is a mainstay of therapy as systemic and urinary alkalinization removes these anions through ion trapping of metabolites. [15]
GIVE IT, along with specific antidotes and possible dialysis
Uremic Acidosis: Uremic acidosis results from the inability of the injured kidney to excrete anions such as phosphates, sulfates, and nitrates and so removal of these substances is the mainstay of therapy. Administration of bicarbonate does not directly impact this end, and data supporting its use is limited. [16] However, it is the current practice of many nephrologists to treat uremic acidosis with bicarbonate infusion to prevent the need for RRT. It is intuitive that bicarbonate can prevent RRT as bicarbonate therapy both corrects pH and also temporarily improves hyperkalemia (depending on the concentration of the solution). This was again demonstrated in the BICAR-ICU trial with a reduced need for RRT in the treatment group, as well as a mortality benefit in a subgroup with AKI. Though further investigation is needed, it is reasonable to give bicarbonate in this population in consultation with nephrology.
GIVE IT, judiciously in severe acidemia and in consultation with a nephrologist
Non-Anion Gap Metabolic Acidosis: In general, this results from loss of total body bicarbonate or retention of additional chloride. It is thus, theoretically reasonable to treat this population with bicarbonate because you are directly addressing the underlying pathophysiology. [1,2]
Renal Losses, including Renal Tubular Acidosis (RTA): Several types exist, but the pathophysiology lies in the inability of the kidneys to re-absorb bicarbonate resulting in increased urinary losses. The mainstay of therapy is bicarbonate, both oral and IV if severe. [17]
GIVE IT
Gastrointestinal Losses (pancreatic fistula, diarrhea, uretal diversion, etc.): Excessive loss of bicarbonate through the GI tract causes a systemic acidosis. Removing the offending pathology (repairing the fistula) is the mainstay of therapy with bicarbonate replacement as a temporizing measure. [18,19]
GIVE IT, in severe cases
Hyperchloremic Metabolic Acidosis: Usually, as the result of iatrogenic over-administration of chloride rich fluids (normal saline). Therapy involves stopping administration of high chloride content fluids and/or switching to a more pH neutral solution such as Lactated Ringer’s or sodium bicarbonate in dextrose. [20,21]
GIVE IT, in severe cases
Conclusions
• Administration of sodium bicarbonate is recommended along with therapies targeting specific etiologies of acidemia in severe cases of non-anion gap metabolic acidosis and anion gap metabolic acidosis secondary to most toxic ingestions.
• Bicarbonate administration is reasonable in severe metabolic acidemia secondary to uremic acidosis and in patients with both AKI and acidemia. Further research is needed to elucidate protocols and to clearly demonstrate benefits.
• Bicarbonate administration is rarely recommended in both ketoacidosis and lactic acidosis unless the patient is in extremis as it has shown no clear benefit and may cause harm.
Expert Commentary
In this trial there was no attempt to differentiate the cause of acidosis a priori, but the type of metabolic acidosis matters when considering bicarb administration. Why?
a) Metabolic acidosis without elevation in the anion gap is more likely to respond to bicarb administration than acidosis with an elevated anion gap. You can think of non-gap acidosis as bicarb deficiency; by administering bicarb, you are repleting bicarb.
b) The trial supports the use of bicarb for uremic acidosis, which tends to be a mix of non-gap- and gap-associated phenomena (renal tubular acidosis combined with an increase in unmeasured anions). Note that the number-needed-to-treat was six patients to prevent one of them from going on dialysis in the AKI subgroup.
c) Lactic acidosis is a misnomer in that the process that creates an elevation in blood lactate anions is physiologically separate from the process generating protons. [1,2] Lactate elevation occurs because of shunting of glycolysis-generated pyruvate away from oxidative metabolism and toward lactate production. This shunting can occur in states of hypoxia (oxidative metabolism shut down, usually Type A) or normoxia (so-called aerobic glycolysis, usually Type B). Lactate is a weak base, so why is there often an associated acidosis? The proton comes from hydrolysis of ATP, which cannot be rapidly replenished under conditions that also favor lactate production (e.g., hypoxia).
So, why does bicarb administration not work well for lactic acidosis? Because even if you titrate off those extra protons using huge amounts of bicarb, you will not rebalance hydrolysis and re-generation of ATP until you fix the underlying problem (ischemia, sepsis, etc.). The rationale for avoiding bicarb in ketoacidosis is similar. Hence, I agree with the recommendation to use bicarb in patients with severe non-uremic anion-gap-associated acidemia only as a temporizing measure while working to reverse the underlying cause.
References
1. Mizock BA. Controversies in lactic acidosis. Implications in critically ill patients. JAMA. 1987;258:497-501.
2. Andersen LW, Mackenhauer J, Roberts JC, Berg KM, Cocchi MN, Donnino MW. Etiology and therapeutic approach to elevated lactate levels. Mayo Clin Proc. 2013;88:1127-1140. PMCID: PMC3975915.

Benjamin Singer, MD
Assistant Professor of Medicine
Pulmonary and Critical Care
Biochemistry and Molecular Genetics
Northwestern University
How To Cite This Post:
[Peer-Reviewed, Web Publication] Jackson, P. Colton, K. (2020, Nov 16). The BICAR-ICU Trial and Practical Use of Bicarb in Metabolic Acidosis. [NUEM Blog. Expert Commentary by Singer, B]. Retrieved from http://www.nuemblog.com/blog/BICAR-ICU-trial.
Other Posts You May Enjoy

References
1. Adams, J, et al. Emergency Medicine: Clinical Essentials. 2013; 2nd edition: 1363-1378.
2. Arbo, J, et al. Decision Making in Emergency Critical Care: An Evidence-Based Handbook. 2015; 1: 496-499.
3. Jaber S, Paugam C, Futier E, et al. Sodium bicarbonate therapy for patients with severe metabolic acidemia in the intensive care unit (BICAR-ICU): a multi-center, open-label, randomized controlled, phase 3 trial. Lancet. June 2018.
4. Berend K, de Vries APJ. Physiological approach to assessment of acid–base disturbances. N Engl J Med 2014; 371: 1434–45.
5. Kraut JA, Madias NE. Lactic acidosis. N Engl J Med 2014; 371: 2309–19.
6. Jung B, Rimmele T, Le Goff C, et al. Severe metabolic or mixed acidemia on intensive care unit admission: incidence, prognosis and administration of buffer therapy: a prospective, multiplecenter study. Crit Care 2011; 15: R238.
7. Cooper DJ, Walley KR, Wiggs BR, Russell JA. Bicarbonate does not improve hemodynamics in critically ill patients who have lactic acidosis. A prospective, controlled clinical study. Ann Intern Med 1990; 112: 492–98.
8. Mathieu D, Neviere R, Billard V, Fleyfel M, Wattel F. Effects of bicarbonate therapy on hemodynamics and tissue oxygenation in patients with lactic acidosis: a prospective, controlled clinical study. Crit Care Med 1991; 19: 1352–56.
9. ElSolh AA, Abou Jaoude P, Porhomayon J. Bicarbonate therapy in the treatment of septic shock: a second look. Intern Emerg Med 2010; 5: 341–47.
10. Kim HJ, Son YK, An WS. Effect of sodium bicarbonate administration on mortality in patients with lactic acidosis: a retrospective analysis. PLoS One 2013; 8: e65283.
11. Gauthier P, Szerlip H. Metabolic acidosis in the intensive care unit. Crit Care Clin. 2002; 18: 289-308.
12. Morris LR, Murphy MB, Kitabchi AE: Bicarbonate therapy in severe diabetic ketoacidosis. Ann Intern Med. 1986; 105: 836-840.
13. Green SM, Rothrock SG, Ho JD, et al.: Failure of adjunctive bicarbonate to improve outcome in severe pediatric diabetic ketoacidosis. Ann Emerg Med. 1998; 31: 41-48
14. Hojer J: Severe metabolic acidosis in the alcoholic: differential diagnosis and management. Hum Exp Toxicol. 1996; 15: 482-488.
15. O’Malley G. Emergency department management of the salicylate-poisoned patient. Emerg Med Clin North Am. 2007; 25(2): 333-346
16. Roderick PJ, Willis NS, Blakeley S, Jones C, Tomson C. Correction of chronic metabolic acidosis for chronic kidney disease patients. Cochrane Database of Systematic Reviews 2007, Issue 1. Art. No.: CD001890. DOI: 10.1002/14651858.CD001890.pub3
17. Morris C, Low J. Metabolic acidosis in the critically ill: Part 2. Causes and treatment. Anesthesia. 2008; 63: 396-411.
18. Callery M, et al. Prevention and management of pancratic fistula. J Gastrointest Surg. 2009; 13(1): 163-173
19. Davidson T, et al. Long-term metabolic and nutritional effects of urinary diversion. Urology. 1995; 46: 804-809.
20. Kellum J. Saline induced hyperchloremic metabolic acidosis. Crit Care Med. 2002; 30: 259-261.
21. Prough D, Bidani A. Hyperchloremic metabolic acidosis is a predictable consequence of intraoperative infusion of 0.9% saline. Anesthesiology. 1999; 90: 1247-1249.
Peripheral Vasopressors: Do I need that central line?

Written by: Saabir Kaskar, MD (NUEM ‘23) Edited by: Abiye Ibiebele, MD (NUEM ‘21) Expert Commentary by: Marc Sala, MD
Vasopressors have been used to treat shock since the early 1900s and continue to remain a mainstay of management of distributive shock. Traditionally, these medicines have been delivered through central venous catheters primarily due to the perceived risks of peripheral infusion, which include potential extravasation of vasoactive medicines and subsequent tissue necrosis. However, central venous catheter insertion is accompanied by its own risks such as pneumothorax, infection and carotid artery insertion and dilation. There is also a risk to delaying vasopressor initiation in hypotensive patients which is why vasopressors are often now started peripherally until central access can be attained.
Peripheral administration of vasopressors has classically been reserved for less potent vasoconstrictors such as phenylephrine and vasopressin. Fear of extravasation and tissue injury often is a cause for concern prior to starting norepinephrine, epinephrine or dopamine peripherally. The perceived harm from administrating these medicines peripherally largely stems from case reports over the past 60 years. However, what does the latest evidence tell us? Is this fear warranted or is it just a myth? Can we send our patients in shock up to the ICUs without central access?
One prospective observational study conducted at Long Island Jewish Medical Center evaluated the safety of vasoactive medication administered through peripheral IV sites. [1] The study monitored the use of vasopressors (norepinephrine, dopamine, and phenylephrine) in an intensive care unit with a total of 734 patients observed. The study incorporated an interdisciplinary protocol between pharmacy, nursing, physicians for administering vasoactive medicines through a peripheral IV. The protocol required that nursing staff examine the PIV access site every two hours, IV size be either 18 or 20 gauge, and utilize upper extremity vein sites with over 4mm vein diameter visualized via ultrasound. During the time of the study, 783 out of 953 patients received vasopressors for 49 +/- 22 hours through peripheral IV. While anatomic position of access site was not formally recorded, most IVs were placed in the upper arm basilic or cephalic vein. Peripheral vasopressors were only allowed to run for 72 hours before running centrally. Of the 783 patients, infiltration of the PIV occurred in 19 (2%) patients. All 19 had prompt local injection of phentolamine and application of nitroglycerin paste at the site of extravasation. No tissue injury was noted at the site of extravasation in any of the 19 cases.
This study shows that administration of vasopressors peripherally is feasible with a low risk if proper precautions are taken. The risk of extravasation and tissue necrosis is still present especially in ED’s and ICUs where such rigorous protocols are not in effect. However, this study demonstrates that vasopressor use may not be an automatic indication for central venous catheter insertion.
A more recent systematic review of peripheral vasopressor safety was recently published in Emergency Medicine Australia. [2] The review incorporated seven observational studies, roughly 1300 patients, that reported the incidence of adverse events for the continuous infusion of peripheral vasopressors including the above study. The major finding was that extravasation events were uncommon (3.4%) and that no significant tissue necrosis or distal ischemia was reported. However, the data analyzed in this review comes from studies with mixed methodology quality and with limited duration of infusion. Five of the seven studies had peripheral vasopressor administration for less than 24 hours.
At its current state, the quality of data reviewing the safety profile of peripheral vasopressors is not universally high. However, the observational data we do have reports low incidence of complications which should be reassuring for clinicians especially when starting these medicines for short periods of time and as a bridge to possible central infusion. Early peripheral infusion should be given more consideration as delaying vasopressor administration has been shown to increase mortality in septic shock. [3] While further research is certainly needed in this field, the current state of data should at least quell some concerns of the perceived risks of peripheral vasopressor administration.
Expert Commentary
Dr. Kaskar does a great job summarizing several of the major studies that can inform how we approach the infusion of peripheral vasoactive drugs in lieu of a central catheter. I can only assume we could agree one area of common ground, which is that if central access is in place, this should be used for the vasoactive infusion, given that the occurrence of tissue complications, while probably rare, can be limb-threatening. An additional prospective study I would mention is by Medlej et al [1] where the authors prospectively studied ED patients managed for a variety of shock states with peripherally administered vasoactive agents and found that 3/55 (5.45% of total, and 6% of those receiving norepinephrine) had extravasation. None had serious complications, but notably among the three events, all three used 20G IVs and two occurred using hand veins. This relatively small and heterogeneous study would indicate that extravasation is uncommon and when it occurs, is not particularly morbid, even with norepinephrine.
Finally, another recent study notable for its cohort took place in the operating room context. Here, medical records of over 14,000 patients who received peripheral norepinephrine to manage hypotension associated with general anesthesia in two European academic centers were studied retrospectively for complications. Only five patients (0.035%) had extravasation, wherein the median infusion duration was 20 minutes, and none of whom had a significant complication from the extravasation. They calculated an estimated a risk of 1-8 events per 10,000 patients.
What do all of these studies have in common that I think belies the true incidence of complications associated with peripheral vasoactive drugs? Vigilance. While it’s true that peripheral norepinephrine infusion may not result in serious tissue necrosis when given in the context of a formal clinical study (especially one that takes place in an operating room with continuous monitoring by anesthesia!), what about when the infusion goes unnoticed during a night shift with a high patient to nurse ratio? In this case, I would argue that the closer to a “real-world experience” we can get with these studies, the better.
I would also mention that a mentor of mine once theorized the “sunset” of crash central lines as the use of intraosseous catheters became more common in adults in the past decade. While intraosseous catheters are not without their own complications, it is worth mentioning their role in this conversation as we move forward in thinking about how to transition patients safely from the ED to ICU with several different options for vascular access in lieu of a controlled, sterile central line placement.
References:
1. Medlej et al. Complications From Administration of Vasopressors Through Peripheral Venous Catheters: An Observational Study. The Journal of Emergency Medicine 2017; 54(1): 47-53.
2. Pancaro et al. Risk of Major Complications After Perioperative Norepinephrine Infusion Through Peripheral Intravenous Lines in a Multicenter Study. Anesthesia and Analgesia 2019; Published ahead of print.

Marc Sala, MD
Assistant Professor of Medicine
Pulmonary and Critical Care
Northwestern University Feinberg School of Medicine
How To Cite This Post:
[Peer-Reviewed, Web Publication] Kaskar, S. Ibiebele, A. (2020, Oct 12). Peripheral Vasopressors: Do I need that central line? [NUEM Blog. Expert Commentary by Sala, M]. Retrieved from http://www.nuemblog.com/blog/abdominal-imaging.
Other Posts You May Enjoy








References
1. Cardenas-Garcia J, Schaub KF, Belchikov YG, Narasimhan M, Koenig SJ, Mayo PH. Safety of peripheral intravenous administration of vasoactive medication. Journal of hospital medicine. 2015; 10(9):581-5
2. Tian DH, Smyth C, Keijzers G, et al. Safety of peripheral administration of vasopressor medications: A systematic review. Emergency medicine Australasia. 2019
3. Beck V, Chateau D, Bryson GL et al. Timing of vasopressor initiation and mortality in septic shock: a cohort study. Crit. Care 2014; 18: R97.
Fluid Responsiveness
Written by: Brett Cohen, MD (NUEM PGY-3) Edited by: Duncan Wilson, MD (NUEM Alum ‘18) Expert commentary by: Luisa Morales-Nebreda, MD
Expert Commentary
I’ll start by saying that the assessment of a patient’s intravascular volume status and fluid responsiveness is one of the most difficult tasks in clinical medicine, yet it remains a crucial one to adequately manage acute circulatory failure.
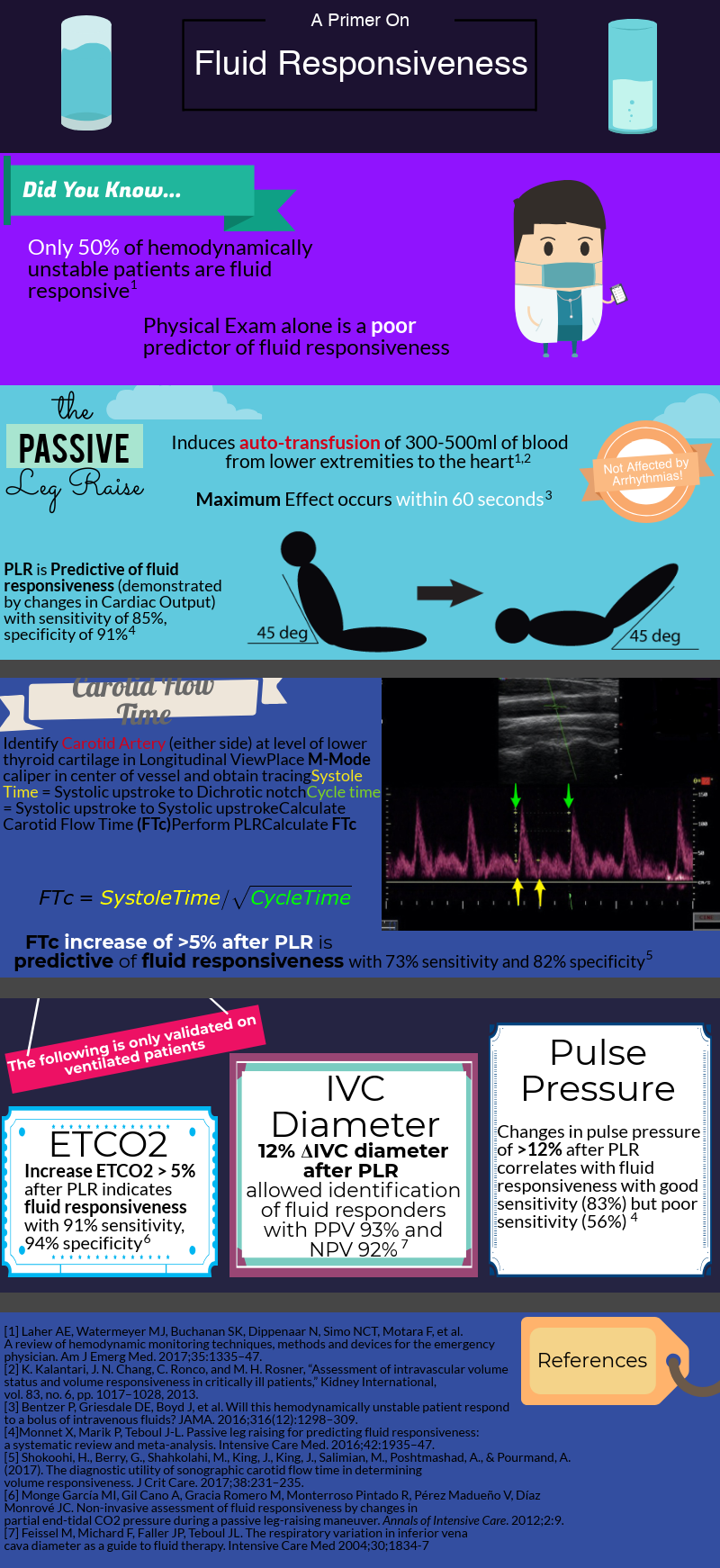
After decades of dedicated research to the critically ill, we’ve learned that rigid protocols lead to large amounts of fluid administration, and that fluid overload is associated with increased morbidity and mortality in septic shock and ARDS patients. As you mentioned, only half of hemodynamically unstable patients are fluid responsive, prompting clinicians to describe novel tools/methods to better evaluate fluid responsiveness.
How do we define fluid responsiveness? What are the determinants of fluid responsiveness?
Fluid responsiveness has been defined as a 10-15% increase in cardiac output after a 500 cc bolus fluid challenge. I find this arbitrary definition unhelpful, but I do think that understanding what determines a fluid bolus leading to a preload-responsive state is important.

Figure 1: Frank Starling curve
When giving a fluid bolus, the expectation is that it will increase cardiac preload (by increasing both the stressed volume and mean circulatory filling pressure). Once this condition has been met, the next assumption is that the increase in venous return will lead to a change in stroke volume/cardiac output (venous return = cardiac output). This is the ideal situation! Both ventricles working on the ascending limb of the Frank-Starling curve without any major changes in other determinants of cardiac output (contractility, afterload, diastolic function) Figure 1. Unfortunately, our critically ill patients are not that simple and most times display significant changes in cardiac contractility (e.g., due to acidosis) and afterload (e.g., due to vasoactive agents) that could place them on the flat portion of this curve.
Cardiac Output = Heart Rate x Stroke Volume
Stroke Volume is determined by: contractility, preload, afterload
How do we measure fluid responsiveness?
Despite a great amount of evidence showing that “static” measurements, such as central venous pressure (CVP) are poor predictors of fluid responsiveness, they continue to be widely used. I don’t mean to say that measuring CVPs is useless. After all, perfusion is determined by the pressure gradient between MAP (mean arterial pressure) and CVP, and CVP is a good marker of preload (just NOT of preload responsiveness).
To circumvent this limitation, we can use “dynamic” measurements of heart-lung interactions during mechanical ventilation. Specifically, if changes in intrathoracic pressure under positive pressure ventilation lead to cyclic changes in stroke volume (stroke volume variation or SVV), pulse pressure (pulse pressure variation PVV), or vena cava diameter it indicates that both ventricles are preload-dependent and the patient is fluid responsive. Limitations to these 3 methods include: a) patients need to be mechanically ventilated and without spontaneous breaths (during which changes in intrathoracic pressure become unreliable) b) tidal volume of at least 8 cc/Kg and normal respiratory system compliance (in order to generate significant swings in intrathoracic pressure, tidal volume needs to be on the high end and your lungs/chest wall can’t be stiff).
So you can think about how limited these maneuvers are in the ICU setting. Most intubated patients take spontaneous breaths (unless paralyzed or deeply sedated). If paralyzed for ARDS, they need to be on low-tidal volume ventilation and their lungs are generally pretty stiff.
As you mentioned a maneuver that is unaffected by these limitations is the passive leg raise test (PLR). Importantly, PLR raises preload by shifting venous blood not only from your lower extremities, but mainly from your splanchnic compartment (where ~70% of unstressed volume reservoir lies). This is why in order to adequately perform the maneuver, patients are placed in a semi-recumbent position (rather than horizontal) and the bed is adjusted to 45֯, followed by assessment of cardiac output changes within 60 seconds (NOT blood pressure changes).
PLR is one of the most validated maneuvers for assessment of fluid responsiveness, and as you described, many downstream methods to measure cardiac output have been used, including: a) velocity time integral of the left ventricular outflow tract b) peak velocity of the carotid artery c) changes in end-tidal CO2.
Given the widespread use of critical care echocardiography and their inherent practicality, I think echocardiographic indices are the most useful dynamic parameters to predict fluid responsiveness.
What to expect and my two cents on fluid responsiveness
Given the pace of technology, innovation in hemodynamic monitoring methods will likely improve in the not too distant future. Pocket echo probes and non-invasive wearable sensors measuring cardiac output will make assessment of fluid responsiveness much easier and reliable. Check out:
Michard et al. Intensive Care Medicine (2017) 43:440-442 https://link.springer.com/article/10.1007%2Fs00134-016-4674-z
Vincent et al. Intensive Care Medicine (2018) 44:922-924 https://link.springer.com/article/10.1007%2Fs00134-018-5205-x
For now, I go to the bedside and after a physical exam perform a basic point-of-care ultrasound to assess heart function, look for interstitial lung edema (B lines) and IVC collapsibility before and after a PLR maneuver (if tolerated). Combining information from these maneuvers not only allows you to better assess a patient’s current volume status and likelihood to be fluid responsive (hyperdynamic LV, absent B lines, collapsible IVC), but can also help you identify what type of shock your patient has and if giving fluid could make things worse (RV failure).
When a patient is likely fluid responsive, I give a small/moderate fluid bolus (250-500 cc) then come back to the bedside to repeat my assessment with the tools I feel most comfortable. Then I ask myself: are things getting better? (Improved mental status/blood pressure, increased in urine output and decreased vasoactive agent requirement). And then do it all over again!

Luisa Morales-Nebreda, MD
Fellow, Pulmonary & Critical Care Medicine
Department of Medicine
Northwestern University
How to Cite this Post
[Peer-Reviewed, Web Publication] Cohen B, Wilson D. (2019, Aug 5). Fluid Responsiveness. [NUEM Blog. Expert Commentary by Morales-Nebreda L]. Retrieved from http://www.nuemblog.com/blog/fluid-responsiveness.